Crystal Directions (The Class Miller)
This section describes the class Miller and gives an overview how to deal with crystal directions in MTEX.
| On this page ... |
| Class Description |
| Defining Miller indices |
| Plotting Miller indices |
| Symmetrically Equivalent Crystal Directions |
| Angle between directions |
| Conversions |
| Calculations |
| Complete Function list |
Class Description
Crystal directions are represented in MTEX by the variables of the class Miller which in turn represent a direction with respect to the crystal coordinate system described by three or four values h, l, k (,m) and a crystall symmetry. Essentially all operations defined for the vector3d class are also available for Miller indices. Furthermore, You can ask for all crystallographically equivalent crystal directions to one Miller index.
Defining Miller indices
Miller indices are defined by three coordinates h, k, l (four in the case of trigonal or hexagonal crystal symmetry) and by the corresponding symmetry class. It is also possible to convert a vector3d object into a Miller index.
cs = crystalSymmetry('trigonal');
m = Miller(1,0,-1,1,cs)
m = Miller(zvector,cs)m = Miller size: 1 x 1 symmetry: -31m, X||a*, Y||b, Z||c* h 1 k 0 i -1 l 1 m = Miller size: 1 x 1 symmetry: -31m, X||a*, Y||b, Z||c* h 0 k 0 i 0 l 1
Plotting Miller indices
Miller indices are plotted as spherical projections. The specific projection as well as whether to plot all equivalent directions can be specified by options.
plot(Miller(2,1,-3,1,cs)) % plot Miller indices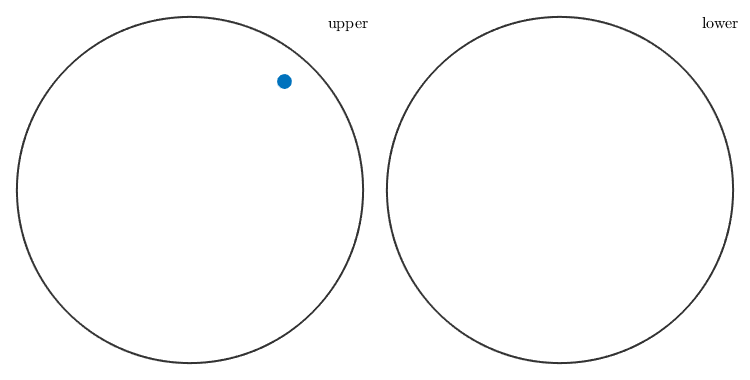
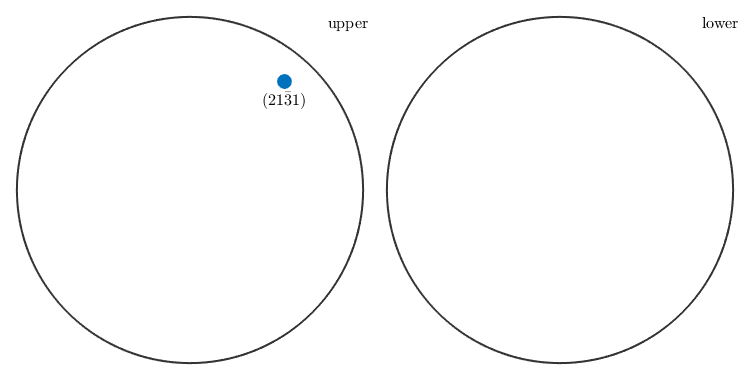
By providing the options all and labeled all symmetrically equivalent crystal directions are plotted together with their correct Miller indices.
plot(Miller(2,1,-3,1,cs),'all','labeled') % plot Miller indices
Symmetrically Equivalent Crystal Directions
A simple way to compute all symmetrically equivalent directions to a given crystal direction is provided by the command symmetrise
m = Miller(1,1,-2,0,cs) symmetrise(m)
m = Miller size: 1 x 1 symmetry: -31m, X||a*, Y||b, Z||c* h 1 k 1 i -2 l 0 ans = Miller size: 6 x 1 symmetry: -31m, X||a*, Y||b, Z||c* h 1 -1 -2 -1 1 2 k 1 -1 1 2 -2 -1 i -2 2 1 -1 1 -1 l 0 0 0 0 0 0
As always the keyword antipodal adds antipodal symmetry to this computation
symmetrise(m,'antipodal')ans = Miller size: 6 x 1 symmetry: -31m, X||a*, Y||b, Z||c* h 1 -1 -2 -1 1 2 k 1 -1 1 2 -2 -1 i -2 2 1 -1 1 -1 l 0 0 0 0 0 0
The command eq or == can be used to check whether two crystal directions are symmetrically equivalent. Compare
Miller(1,1,-2,0,cs) == Miller(-1,-1,2,0,cs)
ans = logical 1
and
eq(Miller(1,1,-2,0,cs),Miller(-1,-1,2,0,cs),'antipodal')ans = logical 1
Angle between directions
The angle between two crystal directions m1 and m2 is defined as the smallest angle between m1 and all symmetrically equivalent directions to m2. This angle in radians is calculated by the function angle
angle(Miller(1,1,-2,0,cs),Miller(-1,-1,2,0,cs)) / degree
ans =
0
As always the keyword antipodal adds antipodal symmetry to this computation
angle(Miller(1,1,-2,0,cs),Miller(-1,-1,2,0,cs),'antipodal') / degreeans =
0
Conversions
Converting Miller indices into a three-dimensional vector is straight forward using the command vectord3d.
vector3d(m)
ans = vector3d
size: 1 x 1
x y z
1.73205 1 0
Conversion into spherical coordinates requires the function polar
[theta,rho] = polar(m)
theta =
1.5708
rho =
0.5236
Calculations
Given a crystal orientation
o = orientation.byEuler(20*degree,30*degree,40*degree,cs)
o = orientation
size: 1 x 1
crystal symmetry : -31m, X||a*, Y||b, Z||c*
specimen symmetry: 1
Bunge Euler angles in degree
phi1 Phi phi2 Inv.
20 30 40 0
one can apply it to a crystal direction to find its coordinates with respect to the specimen coordinate system
o * m
ans = vector3d
size: 1 x 1
x y z
0.0861172 1.76339 0.939693
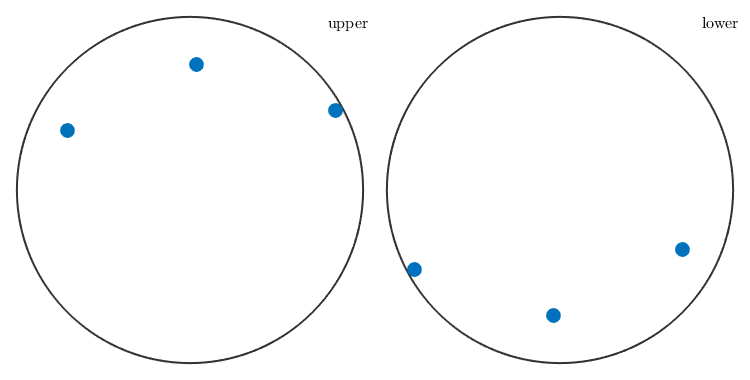
By applying a crystal symmetry class one obtains the coordinates with respect to the specimen coordinate system of all crystallographically equivalent specimen directions.
p = o * symmetrise(m); plot(p)
Complete Function list
| Syntax | |
| m = Miller(h,k,l,cs) | |
| m = Miller(h,k,l,cs,'hkl') | |
| m = Miller(h,k,l,cs,'pole') | |
| m = Miller(h,k,i,l,cs) | |
| m = Miller('(hkl)',cs) | |
| m = Miller(u,v,w,cs,'uvw') | |
| m = Miller(u,v,t,w,cs,'uvw') | |
| m = Miller(u,v,w,cs,'direction') | |
| m = Miller('[uvw]',cs) | |
| m = Miller('[uvw]\[uvw],cs) | |
| m = Miller('(hkl)\(hkl),cs) | |
| m = Miller(x,cs) % transform vector3d to Miller | |
| Input | |
| h,k,l,i(optional) | Miller indice of the plane normal |
| uw,v,w,t(optional) | Miller indice of a direction |
| x | @vector3d |
| cs | crystal @symmetry |
| See also | |
| vector3d_index symmetry_index |
| DocHelp 0.1 beta |